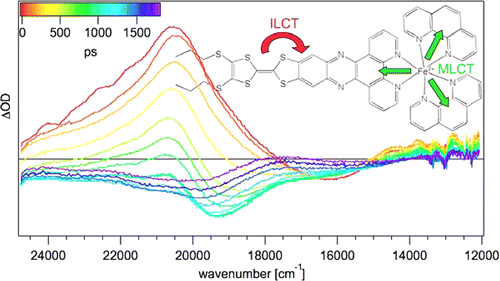
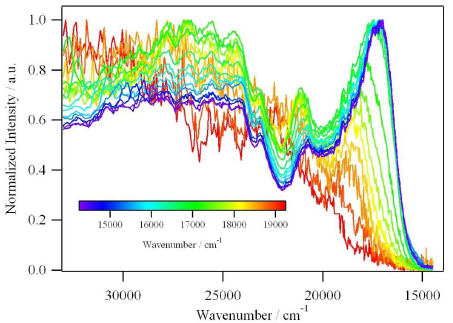
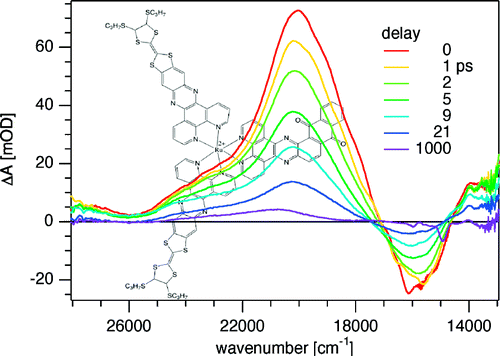
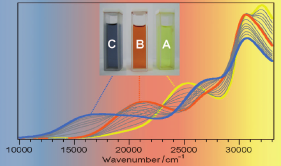
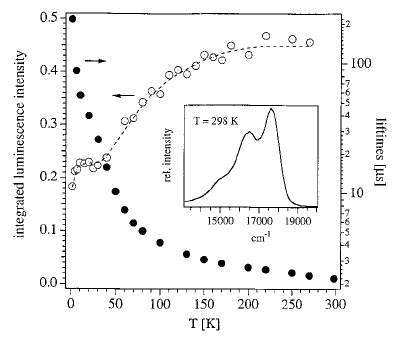
In analogy to the [MII(bpy)3]2+ cations, where MII is a divalent transition-metal and bpy is 2,2â-bipyridine, the tris-chelated [MIII(bpy)3]3+ cations, where MIII is CrIII or CoIII, induce the crystallization of chiral, anionic three-dimensional (3D) coordination polymers of oxalate-bridged (μ-ox) metal complexes with stoichiometries [MII2(ox)3]n2n- or [MIMIII(ox)3]n2n-. The tripositive charge is partially compensated by inclusion of additional complex anions like ClO4-, BF4-, or PF6- which are encapsulated in cubic shaped cavities formed by the bipyridine ligands of the cations. Thus, an elaborate structure of cationic and anionic species within a polymeric anionic network is realized. The compounds isolated and structurally characterized include [CrIII(bpy)3][ClO4] [NaCrIII(ox)3] (1), [CrIII(bpy)3][ClO4][MnII2(ox)3] (2), [CrIII(bpy)3][BF4] [MnII2(ox)3] (3), [CoIII(bpy)3][PF6][NaCrIII(ox)3] (4). Crystal data:  1, cubic, P213, a = 15.523(4) Ã
, Z = 4; 2, cubic, P4132, a = 15.564(3) Ã
, Z = 4; 3, cubic, P4132, a = 15.553(3) Ã
, Z = 4; 4, cubic, P213, a= 15.515(3) Ã
, Z = 4. Furthermore, it seemed likely that 1,2-dithiooxalate (dto) could act as an alternative to the oxalate bridging ligand, and as a result the compound [NiII(phen)3][NaCoIII(dto)3]·C3H6O (5) has successfully been isolated and structurally characterized. Crystal data:  5, orthorhombic, P212121, a = 16.238(4) Ã
, b = 16.225(4) Ã
, c = 18.371(5) Ã
, Z = 4. In addition, the photophysical properties of compound 1 have been investigated in detail. In single crystal absorption spectra of [CrIII(bpy)3][ClO4][NaCrIII(ox)3] (1), the spinâflip transitions of both the [Cr(bpy)3]3+ and the [Cr(ox)3]3- chromophores are observed and can be clearly distinguished. Irradiating into the spin-allowed 4A2 â 4T2absorption band of [Cr(ox)3]3- results in intense luminescence from the 2E state of [Cr(bpy)3]3+as a result of rapid energy transfer processes. |